Патофизиологические основы формирования
неонатального холестаза
Мухина Ю.Г, Дегтярева А.В., Морозов И.А, Туманова Е.Л, Талалаев А.Г.
Кафедра детских болезней № 2 педиатрического факультета с курсом гастроэнтерологии и диетологии ФУВ, Кафедра патологической анатомии педиатрического факультета, РГМУ, ЦНИИГ.
Холестаз – нарушение образования и/или экскреции желчи по желчевыводящей системе, приводящее к внутриклеточному и/или внутрипротоковому накоплению компонентов желчи и проявляющееся желтухой, постоянной или периодической ахолией стула, темным цветом мочи, увеличением размеров печени, кожным зудом, повышением уровня прямой фракции билирубина, щелочной фосфатазы (ЩФ), гамма-глутаминтрансферазы (ГГТ), холестерина, бета-липопротеидов (b -ЛПД) и желчных кислот (ЖК).
В норме секреция желчи зависит от активности ферментных и транспортных систем гепатоцитов и желчных протоков, а также от их структурного и функционального их взаимодействия. Образование первичных ЖК (холевой и хенодезоксихолевой) происходит исключительно в печени из холестерина. Образование вторичных ЖК – дезоксихолевой и литохолевой происходит вследствие 7 a -дегидроксилирования первичных под действием бактерий кишечника. Третичные ЖК и, в основном, урсодезоксихолевая синтезируются в печени путем изомеризации вторичных ЖК. Печеночно-кишечная циркуляция ЖК включает транспорт ЖК в крови, их захват гепатоцитом, транспорт внутри гепатоцита от синусоидального (базолатерального) полюса к канальцевому, конъюгацию деконъюгированных в кишечнике и ре-синтезированных ЖК, экскрецию их в кишечник и последующую реабсорбцию как конъюгированных, так и неконъюгированных ЖК.
Экскреторная функция является жизненно важной; нарушение которой приводит не только к патологическим изменениям гепатобилиарной системы, но и вовлекает в патологический процесс другие органы и системы. Синдром холестаза способствует реализации токсических эффектов различных компонентов желчи, из которых первостепенное значение имеют желчные кислоты, билирубин, холестерин, провоспалительные медиаторы и микроэлементы. Токсические эффекты компонентов желчи представлены в табл. 1.
Таблица 1.
Патологические эффекты компонентов желчи при синдроме холестаза.
|
Компоненты желчи: |
Патологические эффекты: |
|
Желчные кислоты |
Цитотоксичность, нарушение дыхательной цепи митохондрий, апоптоз, повреждение канальцев. |
|
Билирубин |
Нарушение структуры митохондрий |
|
Холестерин |
Повреждение клеточных мембран |
|
Лейкотриены |
Воспаление, гемодинамические эффекты |
|
Медь |
Перекисное окисление липидов |
Дефицит желчи в кишечнике способствует дисрегуляции желудочно-кишечного тракта, нарушению процессов всасывания и, в первую очередь, жиров и жирорастворимых витаминов, нарушению функционального состояния поджелудочной железы. Отставание детей в физическом развитии является характерным признаком хронических холестатических заболеваний гепатобилиарной системы. Вторично формирующийся дисбаланс микроэлементов нарушает процессы оссификации костной системы с развитием остеопороза и метаболических нарушений сердечно-сосудистой системы. Например, при синдроме Алажиля длительно в течение нескольких лет существующий синдром холестаза с постоянно высоким уровнем холестерина и других липидов в крови способствует атеросклеротическим изменениям сосудов уже в детском возрасте. Высокий уровень сывороточных желчных кислот является ведущим фактором в развитии кожного зуда. Основные внепеченочные проявления длительно сохраняющегося синдрома холестаза представлены в таблице 2. Крайний вариант – на фотографии.
Таблица 2.
Внепеченочные проявления синдрома холестаза, фотография ребенка М.
|
Нарушение процессов всасывания |
|
|
Отставание в физическом развитии |
|
|
Признаки дефицита жирорастворимых витаминов: Остеопороз Нейропатия Ксерофтальмия Коагулопатия |
|
|
Кожный зуд |
|
|
Ксантомы, атеросклеротические изменения сосудов. |
У новорожденных и детей первых месяцев жизни синдром холестаза является одним из ранних признаков широкого спектра заболеваний печени и желчевыводящих протоков. В генезе заболеваний лежит несостоятельность тех или иных структур гепатобилиарной системы, являющихся неотъемлемым звеном сложной системы образования и экскреции желчи. Эти дефекты могут быть генетически детерминированными или возникать под действиемэкзогенных факторов. В таблице 3 представлены заболевания гепатобилиарной системы в зависимости от уровня первичного поражения.
Таблица 3.

Первичное поражение клеток печени характерно для большинства гепатотропных инфекционных возбудителей, кроме того может наблюдаться при эндокринных и метаболических нарушениях, а также как проявление токсического действия лекарственных препаратов и осложнение длительного полного парентерального питания.
Гепатит,протекающий с синдромом холестаза, у новорожденных детей может быть вызван широким спектром инфекционных агентов: герпес-вирусом, цитомегаловирусом, энтеровирусами, вирусом краснухи, Эпштейна-Барра, гепатита В и С, возбудителем токсоплазмоза, листериоза, сифилиса и бактериальными агентами. В большинстве случаев неонатальный гепатит, вызванный вышеуказанными возбудителями, является одним из проявлений генерализованной инфекции. Выявление признаков инфекционного процесса и характерного для той или иной инфекции симптомокомплекса является необходимым условием для диагностики гепатита. Исключение составляют вирусные гепатиты В и С, при которых могут отмечаться изменения только печени. Однако у новорожденных детей гепатиты В и С встречаются редко.
Механизм патологического действия включает непосредственное повреждающее действие гепатотропных возбудителей с развитием специфического воспаления гепатобилиарной системы. При неонатальном гепатите синдром холестаза сочетается с биохимическим синдромом цитолиза и нарушением белок-синтетической функции печени. Характерно повышение неспецифических показателей воспаления в общем и биохимическом анализах крови. Наиболее объективным доказательством неонатального гепатита являются результаты морфологического исследования биоптата печени, при котором выявляются типичные для каждой инфекции признаки. Исследования, выявляющие возбудителя и/или антитела к нему подтверждают диагноз.
В настоящее время особенный интерес представляет ЦМВ инфекция. Инфицирование плода на ранних сроках внутриутробного развития может быть одной из причин формирования пороков и/или аномалий развития, в том числе пороков желчевыводящей системы в виде атрезии внепеченочных желчных протоков или гипоплазии внутрипеченочных желчных протоков. При этом ребенок рождается с признаками врожденных пороков и не имеет проявлений гепатита. Нередко встречаются бессимптомные варианты течения, при которых отсутствуют изменения гепатобилиарной системы. В 10-15% случаев клинически не выраженной инфекции могут развиваться поздние проявления, такие как сенсорная глухота, трудности в обучении, а также минимальные мозговые дисфункции. Синдром острой врожденной ЦМВИ (цитомегалия, инклюзионная болезнь) встречается редко и является следствием инфицирования на поздних сроках внутриутробного развития. Типичными для данного синдрома являются: низкая масса тела при рождении, геморрагическая сыпь, тромбоцитопения, анемия, желтуха, гепатоспленомегалия, микроцефалия, нефрит и хориоретинит. Развитие специфического гепатита при этой форме отмечается менее чем в 50% случаев, тогда как чаще выявляется неспецифическая реакция со стороны печени. Диагноз устанавливается на основании выше перечисленного симптомокомплекса, клинико-биохимических признаков гепатита, а также результатов пункционной биопсии печени. Патогномоничным гистологическим признаком гепатита ЦМВ-этиологии является выявление гигантских клеток с ЦМВ включениями - клеток «совиный глаз» (рис. 1).

Рис. 1. Гистологическое исследование биоптата печени при ЦМВ-гепатите.
Среди метаболических заболеваний манифестирующих в течении первых месяцев жизни и проявляющихся синдромом холестаза следует отметить галактоземию, фруктоземию, тирозинемию, болезнь Нимана-Пика (тип С), дефицит-a -1-антитрипсина, неонатальный гемохроматоз и некоторые типы митохондриальной недостаточности с вовлечением ферментов цепи переноса электронов. В основе этих нарушений лежит генетически обусловленный дефект ферментной системы, приводящий к накоплению существующих в норме соединений, а также промежуточных продуктов их метаболизма, обладающих токсическим эффектом на клетки различных органов и являющихся ключевым патогенетическим фактором.
Наиболее часто встречается галактоземия, которая является наследственным заболеванием с аутосомно-рецессивным типом наследования, связанным с 9 хромосомой и включает патологические изменения со стороны печени, почек, ЦНС и глаз. Это заболевание встречается с частотой 1: 50.000 (1:18.000 -1:180.000) живорожденных новорожденных. Первые признаки заболевания появляются в период от нескольких часов до нескольких дней после начала энтерального питания молоком или смесью, содержащей галактозу. В основе этого заболевания лежит дефицит фермента галактоза-1-фосфатуридилтрансферазы, приводящий к накоплению галактозы и галактоза-1-фосфата. Галактоза превращается в галактитол, ответственный за развитие катаракты. Галактоза-1-фосфат значительно снижает синтез энергетических соединений (АТФ, ГТФ, ЦТВ), а также активность ферментов, принимающих участие в глюконеогенезе и синтезе глюкозы из гликогена, вызывая гипогликемию. Эти изменения вызывают тяжелые метаболические нарушения в клетках печени, головного мозга, почек и глаз, способствуют гемолизу эритроцитов. Галактоза-1-фосфат превращается в галактонат и галактонолактон. Эти метаболиты обладают непосредственным гепато-, нефро- и нейротоксическим действием. Описанные изменения лежат в основе характерного для галактоземии симптомокомплекса в виде нарушения общего состояния ребенка, проявляющегося срыгиваниями, рвотой, расстройством стула и др., признаками гемолитической анемии, патологическими изменениями со стороны печени в виде синдрома холестаза, синдрома цитолиза и нарушения синтетической функции. Первым клиническим признаком изменений со стороны ЦНС является внутричерепная гипертензия. Отмечаются также нарушения канальцевой функции почек и формирование катаракты. Подтверждением диагноза служит высокий уровень галактозы в крови, а также дефицит фермента галактоза-1-фосфатуридилтрансферазы.
Характерной особенностью вышеперечисленных метаболических заболеваний также являются патологические изменения со стороны различных органов. Внепеченочные признаки часто предшествуют клинико-лабораторным проявлениям холестаза. Во всех случаях выявляются патологические изменения ЦНС, почек и глаз. У этих больных отмечаются раздражительность, выраженные срыгивания и рвота, плохая прибавка массы тела, частый жидкий стул, клинические эквиваленты гипогликемии и ряд других признаков. При неонатальном гемохроматозе и митохондриальной недостаточности отмечается клиническая симптоматика полиорганной недостаточности. При фруктоземии существует связь между введением в рацион продуктов, содержащих фруктозу или сахарозу и появлением первых признаков заболевания. Особенностью тирозинемии является «капустный запах» и фотофобия, связанная с развитием кератита. При метаболических нарушениях, так же как и, при инфекционных заболеваниях, существует характерный для каждого заболевания симптомокомплекс, играющий ведущую роль в диагностике. Результаты биопсии печени, а также специфические исследования, выявляющие соответствующие дефекты, подтверждают диагноз.
Гистологические признаки имеют особое значение при болезни Нимана-Пика (тип С) и при неонатальном гемохроматозе. При болезни Нимана-Пика типе С вследствие дефицита фермента кислой сфингомиелиназы, участвующей в эстерификации эндогенного холестерина отмечается его накопление в лизосомах макрофагов, что оказывает на них повреждающее действие. При гистологическом исследовании макрофаги, переполненные липидами, выглядят в виде «пенистых» клеток (рис. 2). При неонатальном гемохроматозе диагностическое значение имеет накопление железа в клетках печени, тогда как в норме накопление железа осуществляется в Купферовских клетках.

Рис. 2. Гистологическое исследование биоптата печени при болезни Нимана-Пика, тип С.
Дефицит-a -1-антитрипсина отличается от описанных метаболических нарушений. Единственным его проявлением в неонатальном периоде является синдром холестаза; общее состояние ребенка, как правило, не страдает, патологические изменения в легких развиваются значительно позже, после 3-5 лет жизни. Заболевание имеет аутосомно-рецессивный тип наследования, связанный с нарушением одной аминокислотной цепочки гена, локализующегося в 14 хромосоме.
Конформационные изменения a 1-антитрипсина (a 1-АТ) обуславливают его соединение с соответствующими ферментами. В связи с этим, те или иные структурные нарушения способствуют ослаблению или потере связывающей способности. Фенотипические варианты a 1-АТ отражают скорость его миграции в электрическом поле при pH между 4 и 5. Нормальный белок мигрирует в промежуточной, средней позиции и обозначается М (medium ), белок, мигрирующий более медленно, обозначают Z или S (slow ). В редких случаях отмечается нулевой фенотип - null , при котором полностью отсутствует белок. Наличие двух пар генов предполагает 6 основных вариантов фенотипа, отличающихся степенью выраженности дефицита: MM -100%, MS -75%, MZ -57%, SS -52%, SZ -37%, ZZ -16%. При Z -варианте отмечается замена глутамина на лизин в 342 положении, при S варианте глутамина на валин в 264 положении. Кроме того, при ZZ типе наряду с изменением аминокислотной последовательности имеет место нарушение эндоплазматического ретикулума гепатоцитов, что приводит к нарушению образования боковых углеводородных цепей. В связи с этим только 15% синтезированных полипептидов секретируется в плазму крови, тогда как 85% откладывается в эндоплазматическом ретикулуме, образуя глобулярные включения. Накопление белковых гранул при ZZ фенотипе является ведущим патогенетическим фактором патологических изменений печени. При S варианте отсутствуют патологические изменения гепатобилиарной системы, a -1-АТ не накапливается в клетках печени, однако он имеет нестабильную структуру и подвергается частичному внутриклеточному протеолизу, что обуславливает его сывороточный дефицит. Примерно в 21-50% случаев при неонатальном холестазе, обусловленном дефицитом a 1-АТ по ZZ фенотипу, отмечается развитие ювенильного цирроза печени. Наиболее типичным бронхолегочным проявлением заболевания, встречающимся при любом фенотипе у детей старше 3-5 летнего возраста, является эмфизема легких, однако дефицит a 1-АТ играет важную роль в патогенезе и других хронических заболеваний легких и, прежде всего бронхиальной астмы. Ориентировочным тестом, свидетельствующим о дефиците a -1-антитрипсина, является низкий сывороточный уровень a -1-глобулина. Подтвердить диагноз позволяет низкий уровень a -1-АТ наряду с выявлением характерных гистологических признаков в виде ШИК положительных включений, устойчивых к ферментативному перевариванию и располагающихся преимущественно в перипортальных зонах (рис. 3).

Рис. 3. Гистологическое исследование биоптата печени придефиците a -1-АТ.
Канальцевая мембрана гепатоцита несет на своей поверхности транспортные системы, отвечающие за экскрецию различных компонентов желчи, органических и неорганических веществ, а также большинства ксенобиотиков. В генезе формирования синдрома холестаза играет роль генетически детерминированные дефекты 3-х транспортных систем канальцевой мембраны гепатоцита, лежащих в основе прогрессирующего семейного внутрипеченочного холестаза (ПСВХ) 3-х типов (табл. 4). Н аиболее часто встречается ПСВХ 1-го типа или болезнь Байлера, в основе которой лежит дефицит II -типа АТФ-азы, играющей ключевую роль в экскреции обоих первичных желчных кислот через канальцевую мембрану гепатоцита.При 11 типе ПСВХ отмечается нарушение экскреции преимущественно одной, хенодезоксихолевой кислоты через канальцевую мембрану гепатоцита, связанное с отсутствием на ее поверхности П-гликопротеина.
Таблица 4.
Типы и основные характеристики прогрессирующего семейного внутрипеченочного холестаза.
|
Типы: |
Генный дефект: |
Молекулярные изменения канальцевой мембраны гепатоцита: |
Начальный патогенетический дефект: |
Своеобразие маркеров холестаза: |
|
(болезнь Байлера) |
18q 21 |
Мутация II -типа |
Нарушение экскреции жирорастворимых соединений и в т.ч.желчных кислот |
Низкий уровень ГГТ и холестерина, Повышение ЩФ |
|
(синдром Байлера) |
2q24 |
Отсутствие II -гликопротеина |
Нарушение экскреции преимущественно хенодезоксихолевой кислоты |
Низкий уровень ГГТ и холестерина Повышение ЩФ |
|
III (дефицит MDR -3 гена) |
7q 21.1 |
Отсутствие MDR 3 - II -гликопротеина |
Нарушение экскреции фосфолипидов |
Высокий уровень ГГТ и холестерина Повышение ЩФ |
Примечание: II -тип АТФ-азы - фермент, локализующийся на канальцевой мембране гепатоцита, ответственный за экскрецию преимущественно первичных желчных кислот.
II -гликопротеин - транспортный белок канальцевой мембраны гепатоцита, обеспечивающий
экскрецию, преимущественно хенодезоксихолевой кислоты.
MDR 3 - II -гликопротеин - транспортная система канальцевой мембраны гепатоцита, ответственная за экскрецию фосфолипидов и прежде всего фосфатидилхолина.
Вследствие этих дефектов первичные ЖК накапливаются в клетках печени, а по мере достижения определенной критической концентрации оказывают токсическое действие на гепатоциты и поступают в кровь, способствуя развитию кожного зуда. Неполный вариант синдрома холестаза, имеющий место при этих заболеваниях, проявляющийся нарушением экскреции только ЖК, значительно нарушает процессы кишечного всасывания. Клинические проявления включают синдром холестаза со значительной степенью выраженности кожного зуда, а также отставание детей в физическом развитии и признаки дефицита жирорастворимых витаминов.Характерной лабораторной особенностью I и II типов служит низкий уровень ГГТ и холестерина сыворотки крови наряду с повышением других маркеров холестаза. Фермент ГГТ является мембрано-связанным, локализующимся преимущественно во внутрипеченочных желчных протоках. Основным стимулом для его выделения служат ЖК и, следовательно, заболевания, при которых ЖК не поступают во внутрипеченочную систему, не будут сопровождаться повышением уровня данного фермента. Следует отметить, что в большинстве случаев низкий уровень ГГТ сыворотки крови сочетается с низким уровнем холестерина. Диагностическое значение имеет исследование ЖК, позволяющее определить высокий их уровень в крови и отсутствие или следовые концентрации в желчи.Морфологически выявляется преимущественно внутриклеточное скопление крупных гранул желчи – желчи Байлера (рис. 4).

Рис. 4. Электронно-микроскопическое исследование биоптата печени при болезни Байлера.
В основе III типа ПСВХ (дефицита MDR 3-гена) лежит нарушение экскреции фосфолипидов и, прежде всего фосфатидилхолина, через канальцевую мембрану гепатоцита, связанное с отсутствием на ее поверхности MDR 3-П-гликопротеина. Основным патогенетическим фактором является повреждение внутрипеченочных желчных протоков поступающими в их просвет свободными, несвязанными с фосфолипидами, желчными кислотами. Характерно вовлечение в патологический процесс мелких желчных капилляров, тогда как другие уровни желчевыводящей системы остаются интактными. При гистологическом исследовании определяется пролиферация желчных протоков с постепенным формированием фиброза. При ретроградной или чрезкожной холангиографии изменения не выявляются. Подтверждением диагноза является отсутствие фосфолипидов в желчи и повышенное их содержание в крови.
Важными структурными элементами гепатобилиарной системы являются межклеточные соединения, состоящие из эпителия двух типов: плотного и рыхлого, проницаемого для воды и растворов, обеспечивающие осмотическое равновесие между кровью и желчью, а также межклеточный обмен глюкозой, ионами и аминокислотами (рис. 5а). Межклеточные плотные контакты имеют сложную белковую структуру, схематическое изображение которых представлено на рис. 5б.

Рис. 5 (а). Межклеточные соединения (плотные контакты) в норме (электронная микроскопия)

Рис. 5 (б). Схематичное изображение м ежклеточных плотных контактов.
При развитии холестаза отмечается грубое, неселективное увеличение проницаемости межклеточных соединений, позволяющее осуществлять обратную диффузию канальцевого содержимого, что рассматривается как механизм “предохраняющий” гепатоцит от повреждения. С другой стороны генетически детерминированный дефект межклеточных соединений может быть причиной внутрипеченочного холестаза (рис. 6). Описанный дефект способствует повышенному содержанию компонентов желчи в крови, изменению коллоидных свойств желчи, что приводит к более длительному ее сохранению в клетках печени и внутрипеченочных желчных канальцах.

Рис. 6. Генетически-детерминированный дефект плотных контактов (электронная микроскопия)
Желчевыводящие протоки выполняют важную функцию окончательного формирования желчи, что достигается путем реабсорбции и секреции ее компонентов. В этом процессе задействованы различные транспортные системы и в том числе хлоридные каналы, ответственные за обмен ионов хлора и бикарбоната, а также натрия и водорода, генетический дефект которых лежит в основе муковисцидоза. Нарушение секреции воды, бикарбонатов и других веществ изменяет коллоидное состояние желчи, нарушает ее отток, приводит к длительной обструкции желчных протоков с развитием билиарного цирроза.
Эпителиальные клетки желчных протоков (холангиоциты) отвечают за секрецию биологически активных веществ – IL 6, TGF -β, NO , белка хемотаксиса моноцитов, эндотелина 1, обеспечивая тем самым взаимосвязь с клетками других органов и систем. Возможно, несостоятельность секреторной функции холангиоцитов наряду с генетическими и инфекционными факторами играет определенную роль в формировании хронического воспалительного процесса при первичном склерозирующем холангите. При этом заболевании вовлечение различных участков желчевыводящих путей неодинаковое. Оно может быть ограничено внутри- и внепеченочными желчными протоками. Диагностическими критериями являются четкообразные изменения и стеноз желчных путей, выявляемые при холангиографии (рис. 7а). Гистологически выявляется пролиферация желчных протоков с постепенным формированием вокруг протоков фиброза в виде луковичной шелухи, значительное отложение меди и ступенчатые некрозы (рис. 7б).

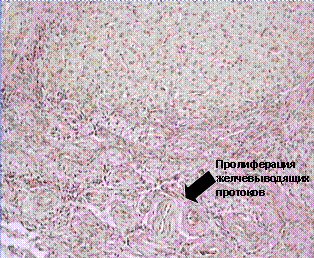
Рис. 7.(а, б). Ретроградная холангиография (а), гистологическое исследование биоптата печени (б) при первичном склерозирующем холангите.
Врожденная гипоплазия внутрипеченочных желчных протоков, проявляющаяся неонатальным холестазом, может сочетаться с аномалиями и/или пороками развития других органов и соответствовать синдрому Алажиля. Диагноз данного синдрома устанавливается на основании выявления двух и более аномалий со стороны других органов (табл. 5).
Таблица 5.
Частота встречаемости диагностических критериев синдрома Алажиля.
Авторы:Основные критерии: |
Emerick et al |
Alagille
|
Deprettere
|
Средний процент |
Синдром внутрипеченочного холестаза |
96% (88/92) |
100%(80/80) |
93% (25/27) |
96% |
|
ВПС (периферический стеноз или гипоплазия легочной артерии) |
||||
|
Офтальмологические изменения (задний эмбриотоксон) |
||||
|
Аномалии развития скелета (расщепление тел позвонков в виде бабочки) |
||||
|
Внутриутробная гипотрофия |
||||
|
Особенностистроения лицевого черепа |
||||
|
Изменения со стороны почек |
||||
|
Дополнительные критерии: |
||||
Нарушения полового развития и высокий голос |
||||
Нейроваскулярные нарушения |
||||
|
Нарушения умственного развития |
Гипоплазия является гистологическим признаком, который оценивается на основании определения отношения желчных протоков к портальным трактам менее 0,6. В норме это отношение должно быть более 0,9.
Выявление гипоплазии внутрипеченочных желчных протоков наряду с отсутствием характерного для синдрома Алажиля симптомокомплекса может свидетельствовать о несиндромальной ее форме. Однако гипоплазия может быть одним из проявлений целого ряда заболеваний и, в том числе хромосомных нарушений (например, синдром Эдвардса), инфекционных заболеваний и некоторых метаболических нарушений. Это диктует необходимость исключения данных заболеваний перед установлением диагноза несиндромальной формы гипоплазии внутрипеченочных желчных протоков.
Атрезия внепеченочных желчных протоков является наиболее частой причиной не онатального холестаза. Патогенез ее можно условно разделить на 2 этапа, первый из которых включает механизмы компенсации, направленные на «сохранение» гепатоцитов. На этом этапе отмечается снижение активности транспортных систем синусоидальной мембраны гепатоцита, что приводит к уменьшению захвата ЖК и других компонентов желчи клетками печени. Отмечается значительное увеличение проницаемости межклеточных соединений, что способствует поступлению желчи из внутрипеченочных желчных протоков в кровь. Изменяется направление внутриклеточного транспорта компонентов желчи и увеличивается проницаемость синусоидальной мембраны гепатоцита для обратного тока желчи из клеток печени в кровь. На втором этапе происходит разрушение внутрипеченочных желчных протоков с формированием характерных ступенчатых некрозов и ихпролиферации, а также деструктивные изменения гепатоцитов с формированием билиарного цирроза печени. Во внутриутробном периоде функцию экскреции и печеночно-кишечной циркуляции выполняет материнский организм, в связи с чем дети с атрезией внепеченочных желчных протоков в большинстве случаев рождаются доношенными и не имеют патологических изменений при рождении.
Первым клиническим признаком является ахолия стула, однако следует помнить о возможности отхождения мекония и, следовательно, появление обесцвеченного стула лишь на 4-5 сутки жизни. Желтуха появляется на 2-3 день жизни, а к началу второй недели кожные проявления уменьшаются. Примерно у 66% больных отмечается развитие «светлого промежутка» в течение 2-х недель. Затем желтуха вновь появляется или нарастает, приобретая зеленоватый оттенок. Характерно увеличение размеров печени к концу 1 месяца жизни, хотя уплотнение ее консистенции может быть выявлено раньше. В более поздние сроки отмечается значительное увеличение и уплотнение печени, постоянная ахолия стула, появляются признаки портальной гипертензии, спленомегалия, кожный зуд и ксантомы. Диагностическое значение имеет сочетание характерных клинических признаков с отсутствием визуализации желчного пузыря при УЗИ. Дополнительное диагностическое значение имеют результаты пункционной биопсии печени и гепатобилиарной сцинтиграфии. Гистологическое исследование биоптата печени выявляет некротически-воспалительную деструкцию как внепеченочных так и внутрипеченочных желчных протоков, признаки билирубино- и желчестаза. Могут выявляться единичные гигантские и полинуклеарные клетки, очаги экстрамедуллярного кроветворения. В последствие отмечается формирование холестаза, портального и перипортального фиброза и цирроза (рис. 8)

Рис. 8. Гистологическое исследование биоптата печени при атрезии внепеченочных желчных протоков.
Киста общего желчного протока в 2-5% случаев вызывает полное нарушение проходимости желчевыводящей системы и, следовательно, может быть причиной внепеченочного холестаза. В большинстве случаев киста сочетается с атрезией внепеченочных желчных протоков. Клинико-лабораторные проявления не отличаются от таковых при атрезии. Диагностическое значение имеет УЗИ, выявляющее полостное образование в проекции общего желчного протока.
Таким образом, развитие синдрома холестаза у новорожденных детей может быть следствием широкого спектра причин с локализацией первичного патогенетического дефекта на разных уровнях гепатобилиарной системы. Патогенетические механизмы различных заболеваний отражают характерный клинико-лабораторный симптомокомплекс, имеющий важное значение в диагностике. Независимо от причины нарушение оттока желчи приводит к реализации токсических эффектов различных ее компонентов и вовлечению в патологический процесс других органов и систем.
Болезнь Байлера
Российский журнал гастроэнтерологии, гепатологии, колопроктологии, 2002, №4, том 12, с.26-30
А. В. Дегтярева, М. И. Пыков, Ю. Г. Мухина, Л. И. Лукина, Н. Ф. Филатова
Гепатоцеллюлярная карцинома (ГЦК) является злокачественной опухолью с быстрым ростом и неблагоприятным прогнозом. Частота ее встречаемости значительно выше у детей с хроническими прогрессирующими болезнями печени, в том числе с болезнью Байлера. В статье представлено клиническое наблюдение - развитие ГЦК у ребенка с болезнью Байлера. Раннее выявление болезни Байлера позволило успешно провести больному родственную трансплантацию печени.
Ключевые слова: гепатоцеллюлярная карцинома, болезнь Байлера, диагностика.
Среди опухолей, поражающих человека, гепато-целлюлярная карцинома (ГЦК) по частоте стоит на 7-м месте. Ежегодно в мире от этой опухоли погибает более миллиона человек . Наиболее часто она встречается у жителей стран Африки и Азии. Однако в последние годы частота случаев ГЦК возрастает и в западных странах, что, вероятно, связано с распространенностью гепатитов В и С, являющихся наиболее частой причиной развития ГЦК.
У детей первичные злокачественные опухоли печени, в том числе и ГЦК, встречаются значительно реже, чем у взрослых. Они составляют около 0,5-2% от числа всех опухолей у детей .
Опыт A.G. Weinberg и M.J. Finegold, включавших 1237 детей с первичными опухолями печени, свидетельствует о следующем их распределении: 1-е место по частоте встречаемости занимает гепатобластома (43%), 2-е - ГЦК (23%), 3-е - рецидивирующие васкулярные опухоли (13%), затем саркомы 6%, мезенхимальные гамартомы (6%), аденомы (2%), фокальная нодулярная гиперплазия (2%) и другие опухоли (5%) . Представленные данные свидетельствуют о том, что примерно в 2/3 случаев первичные опухоли у детей являются злокачественными.
ГЦК является злокачественной опухолью с быстрым ростом и, как правило, c плохим прогнозом. Выживаемость больных ГЦК ниже 25% . Общая классификация включает 2 основных варианта ГЦК: локализованную и с метастазами. Наиболее часто отдаленные метастазы отмечаются в легких, реже - в костях и головном мозге.
Из факторов, предрасполагающих к развитию данной опухоли, у детей первостепенное значение занимают хронические болезни гепатобилиарной системы. Примерно у 30-40% больных, умерших от цирроза печени, при аутопсии выявляется ГЦК .
Группу высокого риска составляют дети с метаболическими нарушениями (тирозинемией, фруктоземией, дефицитом a-1-антитрипсина, наследственным гемохроматозом, гликогенозами и др.), прогрессирующим семейным внутрипеченочным холестазом I (болезнь Байлера) и II типов, хроническими вирусными гепатитами В и С, первичным склерозирующим холангитом и другими болезнями . При биллиарном циррозе вероятность развития ГЦК значительно ниже, чем при других причинах ее развития .
У детей раннего возраста в числе заболеваний, предрасполагающих к развитию ГЦК, ведущее место занимает болезнь Байлера - хроническая прогрессирующая болезнь печени с аутосомно-рециссивным типом наследования. В ее основе лежит генетически детерминированный дефицит мембраносвязанного фермента - АТФазы Р-типа . Этот фермент играет ключевую роль в транспорте жирорастворимых соединений, в том числе желчных кислот (ЖК), через канальцевую мембрану гепатоцита.
Вследствие этого дефекта первичные ЖК накапливаются в клетках печени и оказывают на них повреждающее действие, являясь пусковыми факторами апоптоза. Существует также точка зрения о канцерогенном влиянии избыточного количества первичных ЖК.
Манифестация болезни в виде синдрома холестаза отмечается в первые месяцы жизни, чаще в период новорожденности. Диагностика болезни Байлера основана на выявлении диссоциации между низкой активностью гамма-глутамилтранспептидазы (ГГТП) сыворотки крови, часто в сочетании с низким уровнем холестерина и повышением значения других клинических и лабораторных маркеров холестаза, со значительным повышением уровня ЖК в крови и отсутствием их в желчи, внутриклеточным скоплением желчи (<желчь Байлера>) при электронно-микроскопическом исследовании биоптата печени .
Цирроз печени у детей с болезнью Байлера формируется медленно - в течение 5 лет, а иногда и более. Однако в большинстве случаев больные умирают раньше. Наиболее частой причиной их смерти являются опухоли печени и желчевыводящих протоков. Среди них первое место занимает ГЦК.
Клиническая картина ГЦК полиморфна. Течение болезни может быть бессимптомным. Появление ее первых признаков часто соответствует поздним стадиям. С другой стороны, клинические проявления могут быть настолько яркими, а печеночная недостаточность столь выраженной, что клиническая картина напоминает таковую при абсцессе печени. Спектр проявлений варьирует между этими двумя крайними клиническими формами болезни.
У детей с хроническими болезнями печени, в том числе с болезнью Байлера, развитие ГЦК может сопровождаться нарастанием симптомов основного заболевания, а также увеличением размеров печени. Неспецифическим признаком ГЦК у детей с хроническими болезнями печени являются частые интеркуррентные инфекционные заболевания верхних дыхательных путей и мочевыделительной системы. Это, по-видимому, связано с выработкой a-фетопротеина (a-ФП) и других белков, обладающих выраженным иммунодепрессивным эффектом.
Диагностика ГЦК на ранних стадиях развития в большинстве случаев затруднена. Наиболее простой метод диагностики - ультразвуковое исследование (УЗИ). Выявление эхонегативного образования с нечеткими контурами и неоднородными эхосигналами в печени считается основанием для предположения опухоли и дальнейшего детального обследования больного. Данное исследование позволяет обнаружить очаги поражения диаметром менее 2 см. Допплеровское УЗИ выявляет внутрисосудистое распространение опухоли.
Повышение уровня a-ФП отмечается более чем в 60% случаев . Этот метод имеет важное значение, так как даже невидимые при УЗИ ГЦК могут сопровождаться повышением уровня данного белка.
Однако этот показатель не является специфичным для ГЦК. Он может повышаться при других опухолях, а также при вирусных гепатитах и ряде хронических болезней. Кроме a-ФП, при ГЦК характерно повышение уровня a-1-антитрипсина, кислого a-гликопротеина, дес-g-карбоксипротромбина и активности сывороточной a-L-фукозидазы.
Гематологические изменения, характерные для ГЦК, включают увеличение числа лейкоцитов за счет нейтрофилов. Иногда наблюдается эозинофилия, возможно увеличение количества тромбоцитов. Как правило, нарушается функция свертывающей системы крови. Фибринолитическая активность снижается, что связано с выделением опухолью в сосудистое русло ингибитора фибринолиза.
При компьютерной томографии (КТ) ГЦК выглядит в виде очага пониженной плотности. У больных циррозом печени целесообразно проводить КТ с контрастированием. Йодолипол, введенный в печеночную артерию, быстро выводится из здоровой ткани - в течение 3 нед при фокальной модулярной гиперплазии и значительно дольше - при ГЦК. Введение контрастного вещества позволяет выявить даже мелкие опухолевые очаги диаметром до 2-3 мм.
Магнитно-резонансная томография (МРТ) позволяет получить несколько более четкие изображения, чем при КТ. Внутривенное введение йодсодержащего (соль гадолиния) или магнийсодержащего контрастного вещества повышает эффективность выявления ГЦК.
Селективная ангиография с введением контрастного вещества в чревный ствол или верхнюю брыжеечную артерию позволяет выявить ГЦК, установить ее локализацию, резектабельность и контролировать эффективность лечения.
В большинстве случаев подтвердить диагноз можно на основании данных гистологического исследования биоптата печени. Однако во время этого исследования существует возможность распространения опухоли по ходу иглы .
Клетки опухоли обычно меньшего размера, чем нормальные гепатоциты. Они имеют полигональную форму и зернистую цитоплазму. Имеют неокрашивающуюся, часто пенистую цитоплазму. Иногда обнаруживаются атипичные гигантские клетки. В центре опухоли часто отмечаются очаги некроза. При электронно-микроскопическом исследовании в цитоплазме выявляется гиалин.
Единственный радикальный метод лечения ГЦК - хирургический, заключающийся в резекции опухоли или трансплантации печени. У детей с хроническими прогрессирующими заболеваниями методом выбора служит трансплантация печени. По данным Kaplan-Meier, выживаемость больных с I и II стадиями ГЦК в течение 1 и 5 лет после трансплантации печени составила 91,3 и 72,4% соответственно; у больных с III степенью - 82,4 и 74,1% соответственно (р=0,87) .
В качестве иллюстрации приводим историю болезни ребенка Ш.
Ребенок родился от 30-летней женщины с неотягощенным соматическим анамнезом, от 2-й беременности, протекавшей с угрозой прерывания в I триместре, легким ОРЗ во II триместре; 1-я беременность в 1995 г. закончилась срочными родами (масса при рождении - 2750 г). Однако в возрасте 27 сут жизни ребенок умер. При патологоанатомическом исследовании установлен диагноз гепатита неясной этиологии с формирующимся циррозом печени.
Вторые роды самостоятельные, на 37-38-й неделе беременности. Масса ребенка при рождении - 3000 г, длина - 49 см, состояние - удовлетворительное. В возрасте 4 дней жизни отмечалось нарастание уровня билирубина до 250 мкмоль/л, которое было расценено проявлением конъюгационной желтухи.
Проводилась фототерапия, на фоне которой уровень билирубина снизился, желтуха уменьшилась. Другое лечение в неонатальный период не назначалось.
В возрасте 21 дня отмечены плохая прибавка массы тела, нарастание иктеричности кожного покрова, умеренное увеличение печени до 2,5-3 см ниже реберной дуги по срединно-ключичной линии. Моча приобрела желтый цвет, фекалии периодически обесцвечивались. Патологических изменений в других органах не выявлено. Основные биохимические показатели представлены в таблице.
Результаты биохимических анализов крови больного
Показатель Возраст, мес
0,3* 3 4 5 9 12 19 28** 32*** 46****
Общий белок (57-73) 49 74 65 68 70 64 71 80 73 62
Альбумин (36-49) 38 43 43 44 45 44 51 48 48 44
АлАТ (до 40) 42 3520 1200 220 80 76 91 203 264 32
АсАТ (до 40) 55 2340 1270 190 65 156 153 181 266 14
ЩФ (до 644) 755 786 706 920 720 715 736 542 614 405
Билирубин:
общий (3,4-20,7)
прямой (0,83-3,4)
непрямой (2,56-17,3)
167
42
125
235
150
85
124,8
62,8
62
90
65
25
22
4
18
16,5
3,5
13
19
4
15
99,7
72
27,7
180
120
60
5,0
0
5,0
b-Липопротеиды (1,4-4,5) 3,3 6,9 6,6 - 4,5 4,9 4,7 10 8,6 2,4
Триглицериды (0,39-0,93) - 2,29 1,98 - 1,0 2,1 1,4 2,5 2,5 0,5
Холестерин (1,8-4,9) 3,8 6 5,1 4,5 2,8 3,9 4,1 6 5 3,6
ГГТП (до 50) 281 47 73 30 20 19 17 23 25 14
* 21-й день жизни
** 2 года 4 мес.
*** 2 года 8 мес.
**** 3 года 10 мес (6 мес после трансплантации печени)
1 Норма ГГТП до 1 мес жизни - до 177.
В первом анализе крови отмечалось повышение уровня прямой фракции билирубина и активности щелочной фосфатазы (ЩФ). Значения уровня холестерина и активности ГГТП были низкими.
При последующих анализах нарастало содержание прямой фракции билирубина, b-липопротеидов, триглицеридов, активности ЩФ и аминотрансфераз (значительное). Максимальные значения этих показателей выявлены на 3-м месяце жизни, что совпадало с нарастанием размеров печени до 5,5-6 см ниже реберной дуги. В этом возрасте появился также кожный зуд, который в дальнейшем постепенно усиливался.
Самочувствие ребенка оставалось удовлетворительным. Он активно сосал, не срыгивал. Активность ГГТП была в пределах нормы во всех анализах. Уровень холестерина был низким при большинстве исследований. Значительно повысилось содержание ЖК в сыворотке крови.
При УЗИ выявлялись неспецифические изменения в виде увеличения размеров печени, повышения эхогенности паренхимы. Желчный пузырь имел нормальные размеры и форму.
Дополнительно ребенку проводили трехкратное исследование на маркеры всех вирусных гепатитов, определение аминокислотного спектра сыворотки крови и мочи, уровней галактозы, a-1-антитрипсина крови, антинуклеарных и антимитохондриальных антител. Результаты этих исследований были отрицательными.
При пункционной биопсии печени выявлены признаки преимущественно внутриклеточного холестаза с наличием крупных гранул желчи в гепатоцитах.
Учитывая выявленную диссоциацию между уровнем активности ГГТП и другими показателями холестаза, в том числе ЖК крови, и характерные гистологические изменения, выявленные при биопсии печени, поставлен диагноз болезни Байлера (прогрессирующего семейного внутрипеченочного холестаза I типа).
К возрасту 5-6 мес желтуха постепенно исчезала, нормализовался уровень билирубина, уменьшились размеры печени до +3 см, снизились показатели холестаза и активности ферментов цитолиза. Ведущее место в клинической картине заняли кожный зуд и отставание в физическом развитии.
Дальнейшее динамическое наблюдение (1-2 раза в месяц) ребенка в течение 3 лет жизни свидетельствовало о волнообразном течении болезни с постепенным прогрессированием. Периоды обострения характеризовались нарастанием клинических и лабораторных признаков холестаза и биохимического синдрома холестаза. Белково-синтетическая функция печени не нарушалась.
Снижение протромбинового индекса было связано с нарушением всасывания витамина К. При дополнительном введении препарата витамина К (викасол) он быстро нормализовывался.
Очередное обострение болезни наступило в возрасте 2 лет и 4 мес на фоне острого пиелонефрита. Состояние ребенка ухудшилось. Появилась желтуха, резко увеличились размеры печени до »5-5,5 см, усилился кожный зуд. Фекалии обесцветились, моча стала темной.
На фоне лечения спустя 3 нед клинико-лабораторные признаки пиелонефрита купировались, желтуха несколько уменьшилась, моча посветлела, однако сохранялось увеличение печени.
При УЗИ дополнительных образований не выявлено. Спустя 5 мес ребенку проведено повторное УЗИ, при котором обнаружено опухолевидное образование размерами 30-40 мм в IV сегменте печени.
Допплеровское исследование свидетельствовало о васкуляризации этого образования. Выявлено повышение уровня a-ФП в 16 000 раз. КТ подтвердила наличие опухоли. Пункционную биопсию печени больному не проводили, так как планировалась трансплантация печени.
В возрасте 3 лет и 4 мес в клинике Saint-Luc (Брюссель, Бельгия) ребенку проведена родственная трансплантация печени: пересажена левая доля печени отца. Гистологическое исследование, проведенное после удаления печени, подтвердило наличие ГЦК и болезни Байлера (прогрессирующего семейного внутрипеченочного холестаза I типа). В печеночных лимфоузлах патологических изменений не выявлено.
Послеоперационный период протекал гладко с быстрой нормализацией активности ферментов цитолиза. a-ФП, уровень которого был до трансплантации 117 000 ед., спустя 2,5 нед после трансплантации снизился до 510 ед., а через 2 мес нормализовался (3,5 ед.).
Через 3 мес после трансплантации ребенок возвратился домой. В дальнейшем он наблюдался в отделении катамнеза консультативно-диагностического центра ДКБ № 13 им. Н.Ф. Филатова согласно протоколу, разработанному клиникой Saint-Luc.
К моменту написания статьи (май 2002 г.) катамнез ребенка составил 1,5 года после трансплантации печени. Данные клинического, лабораторного и гистологического исследований свидетельствовали о нормальной структуре и функции гепатобилиарной системы.
Таким образом, ГЦК является злокачественной опухолью с неблагоприятным прогнозом. Частота ее встречаемости значительно выше у детей с хроническими прогрессирующими болезнями печени. Это обстоятельство диктует необходимость трансплантации печени таким больным, не ожидая развития у них цирроза печени.
На этапе ожидания пересадки печени динамическое наблюдение больных должно включать УЗИ и определение уровня a-фетопротеина не реже одного раза в 6 мес. Применение этих методов позволяет выявить данную опухоль на ранних стадиях ее развития, что значительно улучшает прогноз после трансплантации печени.
А.В. Дегтярева (Детская клиническая больница № 13 им. Н.Ф. Филатова)
М.И. Пыков (кафедра лучевой диагностики детского возраста Российской медицинской академии последипломного образования)
Ю.Г. Мухина (кафедра детских болезней № 2 Российского государственного медицинского университета, Москва)
Л.И. Лукина (Детская клиническая больница № 13 им. Н.Ф. Филатова)
| Создан 20 мая 2009 | |||||||||
ОПРЕДЕЛЕНИЕ
Наследственное заболевание, в основе которого лежит нарушение экскреции желчных кислот через канальцевую мембрану гепатоцита. Впервые описано у детей, потомков Джакоба Байлера, и с тех пор названо его именем.
КОД ПО МКБК76.8 - Другие уточненные болезни печени.
ЭТИОЛОГИЯ
Прогрессирующий семейный внутрипеченочный холестаз - следствие генетически-детерминированного нарушения структуры канальцевой мембраны гепатоцита. Имеет аутосомно-рецессивный тип наследования и включает три основных типа: 1тип (болезнь Байлера), II тип (синдром Байлера), III тип (дефицит MDR3 гена). В основе I и II типа лежит нарушение экскреции желчных кислот, тогда как III тип обусловлен нарушением экскреции фосфолипидов.
Ген, ответственный за развитие заболевания, локализуется в регионе длинного плеча 18-й хромосомы (18q21), протяженностью 7сМ в интервале между маркерами D18S69 и D18S64.
ПАТОГЕНЕЗ
В основе болезни Байлера лежит дефицит мембрана-связанного фермента - П-типа АТФ-азы, играющего ключевую роль в транспорте жирорастворимых соединений и желчных кислот через канальцевую мембрану гепатоцита. Вследствие этого дефекта первичные желчные кислоты накапливаются в клетках печени и оказывают на них повреждающее действие, способствуя их разрушению (пусковой фактор апоптоза). С другой стороны, первичные желчные кислоты не поступают в желчную систему и, следовательно, в кишечник, что приводит к нарушению процессов всасывания жиров и жирорастворимых витаминов.
КЛИНИЧЕСКАЯ КАРТИНА
Появление первых признаков холестаза в большинстве случаев отмечают в период новорожденнсти, реже в возрасте 1-10 месяцев жизни. Описаны также случаи желудочно-кишечных и внутричерепных кровотечений, которые предшествовали появлению других клинических признаков болезни. Характерна желтуха, умеренно выраженная гепатомегалия, непостоянная ахолия стула и темный цвет мочи. Типичный признак болезни Байлера - кожный зуд, появляющийся уже в течение первых трех месяцев жизни. Отставание ребенка в физическом развитии и наличие признаков дефицита жирорастворимых витаминов (рахитические изменения и остеопения, мышечная гипотония, сухость кожи и слизистых, тусклость и ломкость ногтей и волос, офтальмоплегия, петехиальная сыпь и/или кровоточивость слизистых) - также характерны для данного заболевания. Синдром холестаза при болезни Байлера имеет волнообразное течение. Факторами, способствующими нарастанию клинико-лабораторных признаков холестаза, служат инфекционные заболевания верхних дыхательных путей и другие интеркурентные заболевания.
ДИАГНОСТИКА
Пренатальиая
Указанные генетические маркеры 18-й хромосомы могут быть также использованы для пренатальной диагностики заболевания и генетического консультирования.
Физикальное исследование
Необходимо оценить цвет кожного покрова и склер, размеры печени и селезенки, цвет стула и мочи. У детей старше трех месяцев жизни может быть кожный зуд.
Лабораторные исследования
Низкий уровень ГГТ и холестерина сыворотки крови наряду с повышением других маркеров холестаза, и в том числе ЩФ, прямой фракции билирубина и желчных кислот.
Характерно повышение ферментов цитолиза и отсутствие изменений белок-синтетической функции печени.
Часто отмечают удлинение ПВ или снижение протромбинового индекса, генез которого связан с нарушением всасывания витамина К в кишечнике.
Для уточнения диагноза при болезни Байлера возможно проведение молекулярно-генетического тестирования специфического локуса длинного плеча 18-й хромосомы (18q21), протяженностью 7сМ в интервале между маркерами D18S69 и D18S64.
Инструментальные исследования
При проведении пункционной биопсии печени отмечают наличие преимущественно внутриклеточного холестаза. Вторично наступает перегруппировка гепатоцитов, образующих тубулярные структуры, псевдоканальца и формирование билирного цирроза печени. Электронная микроскопия обнаруживает желчь в виде грубых гранул («желчь Байлера») в гепатоцитах и внутрипеченочных желчных канальцах.
Дифференциальная диагностика
Проводят с другими заболеваниями печени, проявляющимися внутрипеченочным холестазом с низким уровнем фермента ГГТ (синдром Цельвейгера, нарушение синтеза желчных кислот вследствие ферментопатии).
Показана консультация клинического генетика.
ЛЕЧЕНИЕ
Цели лечения
Коррекция осложнений длительно сохраняющегося холестаза.
Немедикаментозное лечение
Медикаментозное лечение
Урсодезоксихолевая кислота в дозе 20-30 мг/(кгхсут) в 2 приема - постоянно. Жирорастворимые витамины, макро- и микроэлементы (см. лечение АВЖП). У детей более старшего возраста при развитии кожного зуда используют следующие препараты: холестерамин в дозе 4-16 г/день, рифампицин 10 мг/кгхсут) и другие, фототерапия, плазмоферез. Используют также средства, воздействующие на рецепторный аппарат кожи, такие, как ментоловое масло, ланолин, теплые ванны и т.д.
Хирургическое лечение
При развитии патологических состояний, нарушающих качество жизни больного (кожный зуд, отставание в физическом развитии, изменения, обусловленные дефицитом жирорастворимых витаминов), проводят трансплантацию печени.
Дальнейшее ведение
Медикаментозное лечение проводят постоянно. Динамическое амбулаторное обследование 1 раз в 1-2 месяца или по показаниям.
Без трансплантации печени прогноз заболевания неблагоприятный. Больные умирают в возрасте от 2 до 15 лет. Описаны отдельные больные с продолжительностью жизни до 25 лет. Кроме того, по мере прогрессирования заболевания возможно развитие рака печени и желчевыводящей системы.
Прогрессирующий семейный внутрипеченочный холестаз II типа (синдром Байлера)
ОПРЕДЕЛЕНИЕ
Наследственное заболевание, в основе которого лежит нарушение экскреции преимущественно одной первичной желчной кислоты (хенодезоксихолевой), через канальцевую мембрану гепатоцита, связанное с отсутствием на ее поверхности П-гликопротеина.
КОД ПО МКБ-К76.8 - другие уточненные болезни печени.
ЭПИДЕМИОЛОГИЯ
Заболевание встречается в изолированных популяциях на Среднем Востоке. Гренландии и Швеции.
ЭТИОЛОГИЯ
Ген, ответственный за развитие данного заболевания, локализуется во 2-й хромосоме (2q24). Этот ген имеет молекулярную структуру, похожую на таковую гена, ответственного за развитие I типа прогрессирующего семейного внутрипеченочного холестаза, в связи с чем его обозначают «сестринским». В основе прогрессирующего семейного внутрипеченочного холестаза II типа лежит нарушение экскреции преимущественно хенодезоксихолевой кислоты через канальцевую мембрану гепатоцита, связанное с отсутствием на ее поверхности П-гликопротеина.
ПАТОГЕНЕЗ
Не отличается от 1 типа прогрессирующего семейного внутрипеченочного холестаза.
КЛИНИЧЕСКАЯ КАРТИНА
ДИАГНОСТИКА
Пренатальная
Указанные генетические маркеры 2-й хромосомы могут быть использованы для пренатальной диагностики заболевания и генетического консультирования.
Физикальное исследование
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
Лабораторные исследования
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
Дифференциальная диагностика
Проводят с другими заболеваниями печени, проявляющимися внутрипеченочным холестазом с низким уровнем фермента ГГТ. Показана консультация клинического генетика.
Этиопатогенетическое.
Цели лечения: коррекция осложнений длительно сохраняющегося холестаза.
Немедикаментозное лечение
Лечебное питание с повышенным содержанием среднецепочечных триглицеридов.
Медикаментозное лечение
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
Хирургическое лечение
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
Дальнейшее ведение
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
См. «Прогрессирующий семейный внутрипеченочный холестаз I типа».
10 мая 2012 года в Казани состоялась республиканская научно-практическая конференция «Синдром холестаза у детей». Непосредственным организатором конференции выступила кафедра госпитальной педиатрии КГМУ (заведующий кафедрой, профессор В.П. Булатов). Для участия в конференции были приглашены многочисленные педиатры республики Татарстан, неонатологи, врачи смежных специальностей – всего порядка 150 человек. С приветствиями к аудитории обратились главный педиатр МЗ РТ – И.Г. Чигвинцева, профессор А.П. Киясов – проректор по научной и инновационной работе КГМУ, главный врач Детской республиканской клинической больницы МЗ РТ Р.Ф. Шавалиев.
 По словам главного педиатра республики Ирины Григорьевны Чигвинцевой
в последние 20 лет очень многое изменилось в диагностике и лечении заболеваний желчевыводящих путей и печени. «Во многом наш клинический опыт, расширение спектра диагностических исследований позволили выделить из общего класса заболеваний различные нозологии. Синдром холестаза как раз является одним из них. Он встречается достаточно часто среди новорожденных и связан с тяжелыми осложнениями пренатального периода, является следствием врожденной несостоятельности гепатобилиарной системы. В наших условиях, когда мы перешли к выхаживанию детей с экстремально низкой массой тела, проблемы диагностики и своевременное лечение приобретают для нас серьезный актуальный смысл».
По словам главного педиатра республики Ирины Григорьевны Чигвинцевой
в последние 20 лет очень многое изменилось в диагностике и лечении заболеваний желчевыводящих путей и печени. «Во многом наш клинический опыт, расширение спектра диагностических исследований позволили выделить из общего класса заболеваний различные нозологии. Синдром холестаза как раз является одним из них. Он встречается достаточно часто среди новорожденных и связан с тяжелыми осложнениями пренатального периода, является следствием врожденной несостоятельности гепатобилиарной системы. В наших условиях, когда мы перешли к выхаживанию детей с экстремально низкой массой тела, проблемы диагностики и своевременное лечение приобретают для нас серьезный актуальный смысл».
« У новорожденных и детей первых дней жизни синдром холестаза является одним из ранних проявлений заболеваний печени и желчных протоков. Основной предпосылкой являются морфо-функциональные особенности гепатобилиарной системы в этом возрасте. В этом возрасте отмечается относительно низкое количество аминокислот, а также незрелость всех этапов печеночных и кишечных циркуляций. Морфо-функциональные особенности предполагают развитие патологических состояний при определенных условиях», отметила профессор. Под синдромом холестаза принято понимать нарушение образований экскреции желчи по желчевыводящей системе, приводящей к повышению компонентов желчи в крови и недостаточному поступлению в кишечнике. Синдром холестаза имеет характерные клинические и лабораторные проявления. В большинстве случаев синдром холестаза сочетается с синдромом цитолиза повышения активности ферментов трансаминаз, но также и с признаками печеночно-клеточной недостаточности. Клиническими проявлениями холестаза является желтуха, гепатомегалия, ахолия стула, насыщенный цвет мочи. Тактика лечения детей с синдромом холестаза предполагает выявление факторов, способствующих развитию холестаза, затем необходимо исключение заболеваний печени, эффективность лечения которых зависит от сроков его проведения. После этого необходимо адекватное лечение основного заболевания, исключение или ограничение гепатотоксичных лекарств и препаратов крови, максимально раннее начало энтерального питания, также необходимо введение лечебного питания с повышенным содержанием СЦТ, жирорастворимых витаминов через 10 дней, урсодезоксихолиевой кислоты, противопоказано введение гормональных препаратов. По словам профессора, все заболевания желчевыводящих путей принято делить на внепеченочный холестаз и внутрипеченочный холестаз. При внутрипеченочном холестазе визуализируется ахолия стула, у внепеченочного – наоборот. В сообщении докладчик также подробно остановилась на синдроме Байлера. По словам докладчика, дети с таким синдромом составляют группу риска по развитию онкологических заболеваний. Тактика ведения детей с ПВСХ 1 и 2 типов (болезнь Байлера): информирование семьи о том, что возможен риск развития злокачественных новообразований, а также рождения больного ребенка при повторных беременностях. Также, детей с синдромом Байлера необходимо наблюдать один раз в течение двух-трех месяцев. Поддерживающая терапия будет состоять из лечебного питания, использования жирорастворимых витаминов, микро- и макроэлементов, препаратов, уменьшающих кожный зуд. Профессор в рамках сообщения упомянула и о ведении детей с синдромом Алажилль. Прогноз жизни при синдроме Алажилль зависит от течения заболевания, попытки хирургической коррекции ошибочны. Анна Владимировна подробно остановилась и на вопросе диагностики и лечения неонатального гепатита – воспалении печени, вызванном гепатотропными возбудителями: оппортунистическими чаще всего. Участникам конференции были представлены и метаболические нарушения. К ним относятся: тирозинемия, фруктоземия, нарушения синтеза желчных кислот, прогрессирующий семейный внутрипеченочный холестаз, муковисцидоз и другое. Заподозрить такие метаболические нарушения можно, если в семейном анализе присутствуют случаи смерти предыдущих детей от сепсиса с неустановленным возбудителем, синдрома внезапной смерти, неустановленной причины. Помимо этого симптомами метаболических нарушений являются нарушения общего состояния: срыгивания, рвота, потеря веса.
Далее с сообщением выступил заведующий кафедрой госпитальной терапии КГМУ, профессор В.П. Булатову. Он представил доклад «Желчнокаменная болезнь в детском возрасте: дифференцированный подход к терапии». «Желчнокаменная болезнь (ЖКБ) – это болезнь цивилизации», начал свое выступление профессор. Согласно сообщению, частота желчнокаменной болезни (холелитиаза) возросла среди лиц молодого возраста 16-35 лет, при УЗИ новорожденных первых четырех дней желчные камни обнаруживаются в 0,5%. Увеличение роста холелитиаза объясняется улучшением диагностики, а истинный рост заболевания связан с особенностями питания, гиподинамией, ухудшением экологической обстановки. Причинами образования холестериновых желчных камней являются снижение сократительной функции желчного пузыря, перенасыщение желчи холестерином. По словам профессора, билиарный сладж (инородное скопление желчи) формируется у женщин, принимающих оральные контрацептивы и у беременных, а также пациентов, находящихся на парентералом питании или у голодающих. Также к факторам формирования билиарного сладжа относится использование низкокалорийных диет с целью снижения массы тела, а также такие заболевания как: микроцитарная и серповидно-клеточная анемия, цирроз печени, вирусные гепатиты. С 1997 по 2012 год в Детской республиканской клинической больнице МЗ РТ было пролечено 99 больных с ЖКБ, преобладающее число среди них девочки в возрасте 12-14 лет. У 30% больных отмечалась наследственная предрасположенность в основном по материнской линии. Таким больным рекомендуется диета – стол №5. «Это 50% эффективного лечения», отметил профессор. В своем сообщении профессор также выделил показания к консервативной терапии при второй стадии ЖКБ. При обнаружении желчных камней и без отсутствия желчной колики у детей до трех лет рекомендуется только наблюдение. В возрасте от трех до двенадцати лет – операция показана. У детей в подростковом возрасте рекомендуется только выжидательная тактика.
В рамках конференции специалисты также обсудили такие темы как «Синдром холестаза при фетальных гепатитах», были проанализированы причины синдрома холестаза у детей, а также лечение детей с билиарной атрезией.
В завершении конференции участники обменялись мнениями и задали свои вопросы лекторам.
Альфия Хасанова
ПРОЕКТ ПРОТОКОЛА
ДИФФЕРЕНЦИАЛЬНОЙ ДИАГНОСТИКИ И ЛЕЧЕНИЯ СИНДРОМА
ХОЛЕСТАЗА У НОВОРОЖДЕННЫХ ДЕТЕЙ
Введение
Одним из наиболее частых нарушений метаболизма, выявляемых в период новорожденности, является повышение концентрации билирубина в сыворотке крови. При его уровне более 68 мкмоль/л у новорожденных появляется желтушность кожных покровов и склер. Гипербилирубинемия в первые дни жизни может быть обусловлена как физиологическими, так и патологическими причинами и поэтому всегда требует особого внимания. Нарастание интенсивности желтухи, ее зеленоватый оттенок наряду с постепенным увеличением размеров печени, изменением ее консистенции от эластичной до плотной, появление ахолии стула и темного цвета мочи, свидетельствует о нарушении экскреторной функции гепатобилиарной системы – неонатальном холестазе. Лабораторным подтверждением данного синдрома служит повышение прямой фракции билирубина более чем на 15-20% от уровня общего, увеличение концентраций холестерина, бета-липопротеидов (β-ЛПД), желчных кислот (ЖК), а также уровня ферментов щелочной фосфатазы (ЩФ) и гамма-глутаминтрансферазы (ГГТ).
Причинами синдрома холестаза в период новорожденности могут быть морфофункциональные особенности печени и желчных протоков, которые характеризуются высоким уровнем синтеза желчных кислот и незрелостью их печеночно-кишечной циркуляции. Кроме этого, уже на первом месяце жизни могут выявляться первые клинические признаки, свидетельствующие о патологии печени и желчевыводящих протоков, манифестирующих в виде синдрома холестаза.
За последние 20 лет наше понимание основ и патофизиологии многих болезней печени значительно углубилось. Внедрение новых подходов к диагностике и лечению, накопленный клинический опыт позволили установить целый ряд новых нозологических форм, которые до недавнего времени обозначались термином идиопатический неонатальный гепатит. Учитывая что, эффективность лечения врожденных заболеваний печени зависит от сроков его начала, особую значимость приобретает проблема ранней диагностики.
Формирование неонатального холестаза может быть обусловлено заболеваниями гепатобилиарной системы, а также совокупностью неспецифических патологических факторов перинатального периода, т. е иметь внепеченочное происхождение.
Независимо от этиологии холестаза терапия этой категории больных предполагает назначение лечебного питания, дополнительного введения жирорастворимых витаминов , а также использования желчегонного препарата урсодезоксихолевой кислоты. В случае холестаза, обусловленного внепеченочными причинами (транзиторного) на фоне лечения отмечается положительная динамика и постепенное купирование патологического состояния. При болезнях печени и желчывыводящей системы с целью уточнения причины и своевременного назначения этиопатогенетической терапии, дети нуждаются в углубленном обследовании в условиях специализированного отделения.
Неонатальный холестаз, обусловленный внепеченочными причинами.
В структуре внепеченочных причин формирования неонатального холестаза ведущее место занимают состояния, сопровождающиеся развитием гипоксии или ишемии гепатобилиарной системы, гипоперфузией желудочно-кишечного тракта, стойкой гипогликемией, метаболическим ацидозом и застойной сердечно-сосудистой недостаточностью.
Нарушение экскреторной функции гепатобилиарной системы может выявляться у детей с гемолитической болезнью новорожденных на фоне значительного повышения концентрации билирубина. При этом отмечается изменение коллоидных свойств желчи и повышением ее вязкости. В ряде случаев билирубин может оказывать непосредственное токсического действие на мембраны гепатоцитов и митохондрии клеток. Важное место занимают системные и локализованные бактериальные инфекции. Синтез и экскреция сложного каскада медиаторов воспаления купферовскими клетками, а также гепатоцитами и эндотелиальными клетками синусоидов, оказывает непосредственное влияние на образование и экскрецию желчи. Лечебные мероприятия, проводимые новорожденным в условиях отделения реанимации и интенсивной терапии, включают потенциально гепатотоксичные лекарственные средства, полное парентеральное питание, которые также способствуют нарушению функционального состояния гепатобилиарной системы. Развитие холестаза чаще отмечается у недоношенных новорожденных при одновременном действии нескольких патологических и ятрогенных факторов на функцию печени и состояние желчных протоков. В основе этих изменений лежат различной степени выраженности деструктивные изменения желчевыводящих протоков, нарушение проницаемости мембран гепатоцитов и межклеточных соединений, являющиеся в большинстве случаев обратимыми при проведении своевременной терапии. Характерной особенностью неонатального холестаза, обусловленного внепеченочными причинами, является его зависимость от тяжести и длительности патологических состояний перинатального периода и действия ятрогенных факторов. По мере улучшения общего состояния ребенка и разрешения основного заболевания, в большинстве случаев происходит постепенное уменьшение холестаза. Однако остаточные его явления могут сохраняться в течение длительного времени, до 6-8 месяцев жизни. Диагноз неонатального холестаза, являющегося осложнением тяжелой внепеченочной патологии устанавливается при выявлении, предрасполагающих к его развитию факторов и исключения болезней гепатобилиарной системы.
Терапия в этих случаях включает адекватное лечение основного заболевания, ограничение использования потенциально гепатотоксичных лекарств и препаратов крови, Показано назначение патогенетически обоснованной желчегонной терапии препаратом урсодезоксихолевой кислоты (Урсофальк-суспензия), в дозе 15-20 мг/кг/сут. Использование большинства других желчегонных препаратов, содержащих первичные желчные кислоты (холензим, фламин, аллохол и другие) имеет ограничения в неонатальном периоде, т. к. в этом возрасте характерно их высокое образование с проявлением токсического эффекта.
Заболевания печени и желчных протоков, проявляющиеся синдромом холестаза у новорожденных и детей раннего возраста (Таблица 1).
Таблица 1
Заболевания печени и желчных протоков, проявляющиеся синдромом холестаза у новорожденных и детей раннего возраста
Внепеченочный холестаз
Атрезия внепеченочных желчных протоков
Киста общего желчного протока
- "Желчные пробки" и/или камни желчного протока
Сдавление общего желчного протока
Внутрипеченочный холестаз
1. Прогрессирующий семейный внутрипеченочный холестаз (ПСВХ)
1 тип (болезнь Байлера)
11 тип (синдром Байлера)
111 типа(дефицит MDR3 –гена)
2. Доброкачественный семейный внутрипеченочный холестаз
3. Метаболические нарушения
Дефицит альфа-1-антитрипсина
Галактоземия
Фруктоземия
Тирозинемия
Нарушение синтеза желчных кислот, вследствие дефицита ферментов
Пероксисомальная недостаточность (синдром Цельвейгера),
Неонатальный гемохроматоз
Болезнь Нимана-Пика тип С
Митохондриальная недостаточность
4. И нфекционные заболевания (вирусные , бактериальные, вызванные простейшими)
5. Эндокринные нарушения
Гипопитуитаризм
Гипотиреоз
6. Хромосомные нарушения
Трисомия 13, 17 или 18 хромосом
7. Холестаз, связанный с полным парентеральным питанием
8. Холестаз, вызванный токсическим действием лекарственных препаратов
9. Другие
Синдром Алажиля
Несиндромальная форма гипоплазии внутрипеченочных ЖП
Перинатальный склерозирующий холангит
Идиопатический неонатальный гепатит
В зависимости от уровня поражения гепатобилиарной системы принято выделять заболевания, проявляющиеся внепеченочным и внутрипеченочным холестазом, дифференциальная диагностика между которыми основана на сочетании трех признаков: стойкости ахолии стула, уровня ГГТ крови и визуализации желчного пузыря при УЗИ натощак (таблица 2)
Таблица 2.
Дифференциальная диагностика между внепеченочным и внутрипеченочным холестазом у новорожденных детей.
Тип холестаза
Показатели:
Внепеченочный
Холестаз
Внутрипеченочный холестаз
Стойкость ахолии стула
Постоянная
Непостоянная
Уровень ГГТ крови
Визуализация ЖП при УЗИ
Не визуализируется
Визуализируется
ГГТ - гамма глутаминтрансферраза, ЖП – желчный пузырь, УЗИ – ультразвуковое исследование.
Внепеченочный холестаз.
Постоянный характер ахолии стула, повышение сывороточного уровня фермента ГГГ, а также отсутствие визуализации желчного пузыря при УЗИ натощак является характерным признаком внепеченочного холестаза, причинами развития которого в этом возрасте может быть:
1. Атрезия внепеченочных желчных протоков
2. Киста общего желчного протока
3. "Желчные пробки" и/или камни общего желчного протока.
Наиболее частой причиной внепеченочного холестаза и неонатального холестаза в целом является атрезия внепеченочных желчных протоков (АВЖП) , характерным признаком которой служит:
1. В большинстве случаев дети с АВЖП рождаются доношенными с антропометрическими показателями, соответствующими физиологической норме.
2. Желтуха появляется на 2-3 сутки жизни, т. е. в обычные для физиологической желтухи сроки. Примерно у двух третей больных отмечается наличие «светлого промежутка» - уменьшение интенсивности желтухи к концу 1-2 недели жизни с последующим постепенным ее нарастанием и появлением зеленоватого оттенка желтухи к концу 1 месяца.
3. Ахолия стула является наиболее ранним и постоянным клиническим признаком болезни, ее появлению часто предшествует отхождение мекония.
4. Характерным для АВЖП является отсутствие гепатомегалии при рождении с последующим увеличением размеров печени и изменением ее консистенции от эластичной до плотной в течение первых 2 месяцев жизни.
5. К возрасту 1 месяца жизни возможно развитие геморрагического синдрома (кровотечение со слизистых ЖКТ, пупочной ранки, внутричерепное кровоизлияние), обусловленного дефицитом витамин-К зависимых факторов свертываемости крови (ПТИ или ПТВ) в результате нарушения процессов всасывания витамина К в кишечнике.
6. К возрасту 1-2 месяцев жизни, как правило, формируется дефицит веса, степень выраженности которого зависит от вида вскармливания ребенка.
7. Наиболее ранним лабораторным признаком болезни служит повышение билирубина за счет прямой фракции в сыворотке крови, составляющей более 20% от уровня общего билирубина.
8. Характерно повышение других биохимических маркеров холестаза (гамма-глутаминтрансферраза (ГГТ), β-липопротеиды, холестерин, щелочная фосфатаза, желчные кислоты и др.), степень выраженности которых в динамике нарастает от минимального повышения в течение первых 2-3 недель жизни до значительного повышения к 2-3 месяцам.
9. Ферменты цитолиза (АЛТ, АСТ) повышаются умеренно, и, как правило, отсрочено. В большинстве случаев в течение первых 2-3 недель после рождения эти показатели остаются в пределах нормы и затем постепенно повышаются.
10. Показатели, отражающие белково-синтетическую функцию печени (альбумин , фибриноген, ПТИ и др.), на ранних сроках болезни, не изменяются.
11. Информативным методом визуализации гепатобилиарной системы является УЗИ, при проведении которого желчный пузырь натощак не визуализируется или выявляется в виде «гиперэхогенного тяжа». В ряде случаев при атрезии желчных протоков выявляют расширение внутрипеченочных желчных протоков, реже - кисты в воротах печени и полисплению.
12. Дополнительное диагностическое значение имеют гепатобилиарная сцинтиграфия, ретроградная холецистохолангиография (РХПГ), магниторезонансная томография (МРТ) и биопсия печени. При гепатобилиарной сцинтиграфии, имеющей достаточно высокую чувствительность и специфичность у больных с АВЖП отмечается отсутствие поступления радиоизотопного вещества в кишечник наряду с удовлетворительной поглотительной и накопительной функцией печени. Проведение РХПГ имеет целый ряд технических ограничений у детей первых месяцев жизни.
13. Морфологическое исследование биоптата печени показывает различной степени выраженности сгустки желчи, пролиферацию желчных протоков, отек портальных трактов и фиброз.
Диагноз галактоземии устанавливают на основании повышенного содержания галактозы в сыворотке крови, редуцирующих веществ в моче, дефиците фермента галактоза-1-фосфат уридилтрансферразы в эритроцитах, лейкоцитах и гепатоцитах. Возможно также генетическое тестирование специфического локуса.
Основным методом лечения является диета с полным исключением галактозы и лактозы (Прегестимил, Нутрамиген, АL110 и другие). Своевременное начало лечебного питания приводит к полному выздоровлению больного. Контроль за эффективностью лечебного питания целесообразно проводить путем определения уровня галактоза - 1 - фосфата в эритроцитах. Допустимым считается повышение данного метаболита до 3 мг/дл.
При своевременном назначении лечебного питания прогноз благоприятный. Описаны редкие случаи развития цирроза печени и/или печеночно-клеточной карциномы. Может также отмечаться низкий интеллектуальный индекс с нарушением абстрактного мышления, речи и зрительного восприятия, мышечная гипотония, тремор, атаксия и умственная отсталость. Кроме того, описаны случаи нарушения репродуктивной функции у женщин с формированием гипергонадотропного гипогонадизма.
Эндокринные нарушения. Синдром холестаза является одним из проявлений гипотиреоза или гипопитуитаризма, которые имеют типичные клинико-лабораторные проявления. Низкий уровень соответствующих гормонов подтверждает диагноз. Врожденные эндокринные нарушения служат показанием к проведению заместительной гормональной терапии.
Токсическое действие лекарств. В большинстве случаев при холестазе, вызванном токсическим действием лекарственных препаратов существует указание об использовании потенциально гепатотоксичного лекарства, что имеет определенное значение в диагностике. К потенциально гепатотоксичным препаратам могут быть отнесены некоторые антибиотики (тетрациклин, эритромицин, линкомицин, новобиоцин, клавулановая кислота, ампициллин, левомицетин, гентамицин, цефалоспорины 1 поколения, тиенам), мочегонные (лазикс), нестероидные противовоспалительные (индометецин), нитрофураны (фурагин, 5-нок), сульфониламидные препараты, антиконвульксанты и нейролептики. Лечение токсического поражения печени включает исключение потенциально гепатотоксичных лекарств, желчегонную и посиндромную терапию.
Длительное полное парентеральное питание. Холестаз, обусловленный длительным полным парентеральным питанием (ППП) может быть диагностирован у новорожденных, получающих ППП более 2-х недель и у детей более старшего возраста – более 3-4 недель. После начала энтерального питания характерно уменьшение признаков холестаза. Наиболее эффективным этиопатогенетическим лечением при этом является максимально раннее начало энтерального питания, а также желчегонная терапия.
Идиопатический неонатальный гепатит . Диагноз идиопатического неонатального гепатита устанавливается при исключении всех известных причин развития синдрома холестаза и выявлении гигантоклеточной трансформации гепатоцитов при гистологическом исследовании биоптата печени.
Характерной особенностью заболеваний с преимущественным поражением внутрипеченочных желчных протоков, а также дефицита-a-1-антитрипсина является удовлетворительное самочувствие больных и отсутствие патологических изменений других органов и систем (таблица 3). Дефицит-a-1-антитрипсина в течение первого месяца жизни проявляется синдромом холестаза, тогда как патологические изменения со стороны легких развиваются значительно позже, после 3-5 лет жизни. Диагностическое значение имеет низкий сывороточный уровень альфа-1-антитрипсина и альфа-1-глобулина.
В эту подгруппу синдром Алажиля отнесен условно. Для него характерны аномалии и/или пороки развития других органов, однако в большинстве случаев они не имеют клинического значения. Диагностика синдрома Алажиля основана на выявлении характерных особенностей фенотипа и 2-х или более типичных аномалий и/или пороков развития других органов:
1. Выявление признаков внутриутробной гипотрофии (весо-ростовой показатель для доношенных новорожденных, соответствие веса при рождении гестационному возрасту при преждевременных родах).
2. Выявление фенотипических особенностей (широкий, выступающий лоб, гипоплазия средней трети лица, глубокопосаженные, широкорасставленные глаза (гипертелоризм), длинный прямой нос с утолщением на кончике, выступающий подбородок, редкая дерматоглифика и др.)
3. ЭХО-КГ (периферический стеноз или гипоплазия легочной артерии)
4. Консультация Офтальмолога (задний или передний эмбриотоксон)
5. Рентген позвоночника – прямая проэкция (расщепление тел позвонков в виде «бабочки».
Реже при данном синдроме встречаются аномалии мочевыделительной системы и других органов.
Для подтверждения диагноза проводится пункционная биопсия печени, при которой выявляют гипоплазию внутрипеченочных желчных протоков. Этиопатогенетическое лечение данного синдрома отсутствует. При формировании цирроза печени и отсутствии грубых пороков сердца или почек единственным радикальным методом лечения является трансплантации печени.
ПСВХ 3 типа
(дефицит MDR3 гена)
Отсутствие фосфолипидов в желче
ПСВХ 3 типа - прогрессирующий семейный внутрипеченочный холестаз, MDR3 ген – ген полилекарственной резистентности, ж. п. – желчные протоки.
Выявление гипоплазии внутрипеченочных желчных протоков (при отсутствии характерных для синдрома Алажиля признаков) свидетельствует о несиндромальной форме гипоплазии внутрипеченочных желчных протоков. Обнаружение пролиферации желчных протоков требует дополнительного проведения ретроградной или чрезкожной холангиографии. Деформация желчных протоков, выявляемая при холангиографии в сочетании с пролиферацией желчных протоков при гистологическом исследовании биоптата печени позволяет установить диагноз перинатального склерозирующего холангита. Отсутствие изменений при этом исследовании свидетельствует о прогрессирующем семейном внутрипеченочном холестазе (ПСВХ) 3 типа, подтверждением которого является отсутствие фосфолипидов в желче.
Симптоматическое лечение синдрома холестаза
Симптоматическое лечение направлено на профилактику и лечение осложнений длительно сохраняющегося синдрома холестаза и формирующегося цирроза печени
При всех заболеваниях, проявляющихся синдромом холестаза за исключением атрезии внепеченочных желчных протоков, кисты общего желчного протока и нарушения синтеза желчных кислот, вследствие ферментопатии, показано проведение желчегонной терапии препаратом урсодезоксихолевой кислоты (Урсофальк). Использование других желчегонных препаратов имеет ограничения у детей первых месяцев жизни, так как многие из них (холензим, фламин и другие) содержат высушенную желчь крупного рогатого скота и, следовательно первичные ЖК, образование которых в этом возрасте и так велико. Урсодезоксихолевая кислота (УДХК) представляет собой нетоксичную третичную желчную кислоту (ЖК), в норме содержащуюся в желчи человека в небольших количествах (не более 5% от общего пула желчных кислот). Она является более полярной и гидрофильной по сравнению с другими ЖК. Эти свойства обуславливают практически полное отсутствие токсичности данного соединения, а также ее высокую холекинетическую активность. Спектр терапевтического действия УДХК включает несколько основных механизмов:
1. УДХК образует смешанные мицеллы с неполярными гидрофобными ЖК, что значительно уменьшает потенциал их токсичности и усиливает их экзоцитоз путем активации кальций-зависимой альфа-протеинкиназы.
2. УДХК индуцирует холерез богатый бикарбонатами, что приводит к увеличению пассажа желчи и стимулирует выведение токсичных ЖК и билирубина.
3. Неполярные димеры этой ЖК встраиваются во внутренний слой мембран гепатоцитов, стабилизируя их структуру.
4. УДХК обладает способностью уменьшать всасывание токсичных и липофильных ЖК и других компонентов желчи в подвздошной кишке.
5. УДХК снижает экспрессию антигенов главного комплекса гистосовместимости (HLA 1 класса на гепатоцитах и HLA 2 класса на эпителиальных клетках желчных протоков), которые обуславливают активацию цитотоксичных Т-лимфоцитов.
Препараты урсодезоксихолевой кислоты назначаются из расчета 15-20 мг/кг/сут. При недостаточной эффективности доза может быть увеличена до 30 мг/кг/сут. При проведении длительного лечения более 1-2 месяцев используется поддерживающая доза 10 мг/кг/сут.
Для обеспечения нормального темпа роста таким детям требуется увеличение белковой и калорийной нагрузки по сравнению со здоровыми детьми а, также содержание в диете среднецепочечных триглицеридов (СЦТ). При этом важным условием является сбалансированность рациона питания. Это может быть достигнуто путем использования в рационе специальной лечебной диеты. Если это не приводит к желаемому эффекту, питание проводится через назогастральный зонд или осуществляется частичное парентеральное питание.
При наличии грудного молока и отсутствии противопоказаний к его приему у ребенка, следует назначать ферментные препараты (креон – 1тыс ЕД/кг/сут) или комбинировать грудное молоко с лечебной смесью, содержащей СЦТ.
При отсутствии грудного вскармливания, потребность детей в основных пищевых ингредиентах может быть достигнута при использовании лечебного питания, содержащего СЦТ или проведении частичного парентерального питания.
Необходимое для детей с синдромом холестаза количество СЦТ (50-60%) содержится в лечебной смесе Хумана, СЦТ, а также в смесях на основе белковых гидролизатов (Прегестимил, Алфаре и др.). Использование последних не вполне оправдано у этой категории больных, т. к. в большинстве случаев у них нет нарушений всасывания белка и аллергии к нему, а эти смеси имеют значительно худшие вкусовые качества и более высокую стоимость. Наш опыт использования смеси Хумана, СЦТ у детей с хроническими прогрессирующими заболеваниями печени свидетельствует о хорошей ее переносимости и значительном улучшении нутритивного статуса больных.
Важным компонентом питания являются жирорастворимые витамины и микроэлементы, дефицит которых отмечается у всех больных с длительно сохраняющимся синдромом холестаза. Рекомендуемые дозы жирорастворимых витаминов и микроэлементов представлены в таблице 6 .
Таблица 6.
с синдромом холестаза.
Название:
Доза:
Путь введения:
Кратность:
Витамин Д
30000 – 60000 МЕ
*5.000 – 8.000 МЕ
1 раз/мес.
1раз/сут
Витамин А
25000 – 50000 МЕ
*5.000-20.000 МЕ
1 раз/мес.
1 раз/сут
Витамин Е
*25 МЕ/кг/день
peros (tocopherol polyethylene glycol 1000 succinate)
1раз/2 нед.
Витамин К (викасол)
1мг/кг (мах–10 мг)
1раз/1-2 нед.
1раз/день
Цинк(цинк сульфат)
1 раз/день
*при отсутствии мониторинга уровня витаминов в крови следует отдавать предпочтение пероральному пути введения.
** из рассчета элементарного Са, отношение Са/Ф = 2.
Большинство хронических заболеваний печени, проявляющихся синдромом холестаза, сопровождаются кожным зудом, значительно нарушающим качество жизни больных. Существуют разные методы, уменьшающие кожный зуд. Это лекарственные препараты с различным механизмом действия (холестерамин, рифампицин, урсофальк, гептрал и др.), фототерапия, плазмоферез и операция билиарного отведения. Используются также средства, воздействующие на рецепторный аппарат кожи, такие как ментоловое масло, ланолин, теплые ванны и т. д.
Осложнениями цирроза печени являются портальная гипертензия, гепаторенальный и гепатопульмональный синдромы, бактериальный перитонит и/или холангит, а также печеночная энцефалопатия. Лечение портальной гипертензии включает ограничение соли, использование мочегонных препаратов, восполнение уровня альбумина в крови, а в тяжелых случаях - парацентез. Расширенные вены пищевода и желудка являются показанием к назначению Н2 –блокаторов и, в ряде случаев осуществлению склеротерапии. Формирование гепаторенального и гепатопульмонального синдромов является абсолютным показанием к трансплантации печени. При бактериальных осложнениях осуществляется антибактериальная терапия. Появление признаков энцефалопатии служит основанием для назначения диеты с низким содержанием белка и применения препаратов лактулозы (Дюфалак и др.).
Таким образом, неонатальный холестаз является одним из ранних признаков широкого спектра болезней печени и желчных протоков, а также может иметь внепеченочное происхождение. Эффективность лечения многих наследственных и врожденных заболеваний гепатобилиарной системы зависит от сроков ее начала, и следовательно, определяет необходимость их ранней диагностики. Алгоритм дифференциальной диагностики предусматривает оптимальный перечень исследований, необходимый для установления диагноза в максимально короткие сроки.